Early-stage development
The development and commercialization of the products of biotechnology innovation require complicated, elaborate and therefore lengthy and costly R&D efforts as well as complex manufacturing capabilities.
Manufacturing facilities for the production of biotechnology-derived medicines, as well as the drugs themselves, are subject to strict licensing requirements by regulatory authorities around the world. Complex regulatory approval processes apply in particular to biosimilars, the successor products of biopharmaceutical drugs, which – unlike generics as successor products of chemical-synthetic drugs – cannot be reproduced identically.
A similar complexity is also evident in other areas of biotechnological innovation. When products such as genetically modified crops or livestock or biocontrol agents, as well as some foods or industrial enzymes, made through biotechnological processes are released into the environment, they are subject to strict environmental regulations. Similarly, in most jurisdictions, biotechnologically modified plant varieties that might be used in plant breeding need to comply with phytosanitary measures and other market approval and registration requirements, as set out in seed laws for all seeds entering the market. Finally, foods based on genetically modified crops have to meet strict requirements set out by environmental and food laws.
Such regulatory requirements differ significantly among jurisdictions and, in practice, may interact with the exercise of IP rights by the innovators at the technology transfer and commercialization stages of those biotechnological innovations in those jurisdictions. While they are not addressed in this Primer, it should be noted that, in the life sciences field, market approval requirements additionally make the technology transfer and commercialization of biotechnological products and processes time and resource intensive. In short, research in biotechnology is very capital intensive, takes longer to mature than other fields of technology, and the manufacture of innovative products is often more expensive when compared with other domains.
To facilitate understanding of what it takes to bring a biotechnology product to market, this section of the Primer traces a biotech medicine from the start of R&D activities to the moment an active ingredient is discovered, through to testing and application for market approval by the competent authorities, and on to market approval and use in the patient who needs it, and finally to post-marketing surveillance obligations once the product is on the market. Oftentimes it is the discovery of a gene or a gene product (usually a protein) with a unique property that ultimately leads to a commercial product. This discovery can take place in a public sector institution such as an academic research laboratory, or in a private-sector laboratory. The example presented here considers a discovery in an academic research laboratory that is likely to be the recipient of public funding.
Jurisdictional divergences
In many countries, research arising from public funding is governed by specific regulations designed to protect the public interest. These regulations set out specific requirements that must be met by the recipient of such funding. The subject matter, the conditions for patentability and the ownership of an innovation may differ from jurisdiction to jurisdiction.
The United States of America
In the United States of America (US), the transfer of technology developed from publicly funded research (research funded by federal grants to an academic research laboratory or nonprofit research institution, for example) to private entities for commercialization is governed by the Bayh-Dole legislation.
European Union
For example, in the European Union there is no legislation at an EU level. Regulation therefore differs between EU member states. With regard to technology transfer agreements, the EU Technology Transfer Block Exemption Regulation (TTBER)
The United Kingdom
In the United Kingdom (UK), although there is no specific legislation as such, the 1977 Patents Act, which harmonized UK law with the European Patent Convention, makes clear that inventions belong to inventors unless made in the normal course of their duties to their employers, in which case the invention belongs to their employer.
Germany
In Germany, an employee invention is a patentable or utility model invention made by an employee in the course of his or her official duties. Under the German Employee Inventions Act, the employer is generally entitled to the rights to the service invention, whereas the employee only has a compensatory claim to remuneration. Even after the abolition of the so-called university lecturer privilege, special provisions apply to inventions made by employees at a university. The law also regulates the treatment of creative achievements by employees that cannot be protected by a patent or utility model or other means, but which improve the performance of a company (“technical improvement proposals”).
Nevertheless, in an ideal setting, after many years of R&D, a researcher develops an invention
Marketing authorization
In the marketing authorization procedure for a pharmaceutical product, the quality, efficacy and safety of the product must be tested before it can be administered to patients. In the context of efficacy, the medicinal product is assessed to ensure it has the described effect for the indication in question, while with regard to safety, any side effects associated with the product are evaluated. To enable the regulatory authorities to verify quality, safety and efficacy, the applicant must submit the results of preclinical and clinical studies with the application for marketing authorization.
Patent application licensing
When patent rights are licensed before a patent is granted, the license agreement will often address unique issues that may arise during the term of the agreement, including which party has the right to lead and make decisions regarding prosecution of the patent applications, what happens if the parties disagree whether to pursue or maintain a patent application filing, who is responsible for the patenting costs, and whether the licensee is required to pay earned royalties on pending claims of a patent application. If a patent application is not licensed within a few years of filing, the patent holder may decide to abandon the patent application because of its weakened ability to attract investment over time considering the ever-shortening patent life. Ongoing acceleration of innovation and shortening of product life cycles in multiple biotechnology sectors may further contribute to such abandonment decisions. As well as the time required, the high costs of building global patent portfolios may be a challenge for patent applicants.
Besides these practical considerations, the combination of increasingly data-driven incremental innovation in the delivery of biological services, such as genome sequencing or editing, together with raised eligibility requirements in multiple jurisdictions for certain biotechnology patents, particularly “gene” patents claiming complementary DNA (cDNA) sequences, for example, for diagnostic tools, further contribute to changing trends in the frequency and extent of gene patenting in various biotechnology sectors. As the role of patents therefore changes within IP and technology transfer practices in the life sciences, biotech innovators are seeking to develop new tools and arrangements to facilitate efficient technology transfer within evolving life science ecosystems. Box 3.1 presents an overview of the early stages of moving biotechnological inventions from bench to market.

Study of multiple active ingredients in laboratory (screening)
Discovery in laboratory (public or private)
Extensive research for conception of a patentable invention
Patented within one year of discovery
Technology Transfer Office in university
Supplementary protection certificates (SPCs)
Supplementary protection certificates (SPCs) are an IP right that serve to extend patent rights, particularly protection of the reward for the investments made to obtain the invention. They apply to specific pharmaceutical and plant protection products that have been authorized by regulatory authorities and aim to offset the loss of patent protection for such products that occurs because of the compulsory lengthy testing and clinical trials they require before obtaining regulatory marketing approval.
The European Union, for example, wishes to provide sufficient protection for these products in the interest of public health and to encourage innovation in these areas to generate smart growth and jobs. An SPC can extend a patent right for a maximum of five years. A six-month additional extension is available in accordance with Regulation (EC) No 1901/2006
Exclusive licenses
Additionally, biotech patents are often licensed on an exclusive basis to provide the licensee with adequate financial incentive to invest in further developing the market potential of the technology. An exclusive license agreement is one of the most commonly used agreements to facilitate the transfer of an innovation to the marketplace. It both supports the licensee’s ability to seek outside investment (or to justify to its shareholders the expenditure of internal resources to develop the technology) and have adequate autonomy over commercial development, while at the same time providing the patent owner with some degree of control over its IP assets to ensure it will have the right to participate in the licensee’s prospective financial success.
Nonexclusive licenses
However, in the case of some platform technologies (such as technologies that could be exploited in multiple fields of use) the licensing strategy may be instead to pursue as many nonexclusive licenses as possible (assuming such a strategy is palatable to prospective licensees), or to require the exclusive licensee to grant sublicenses in fields of use that it is not developing and/or that do not interfere with its development plans (in cases where nonexclusive licensing is not a viable strategy given low industry interest). This is particularly appropriate where a licensee cannot cover the whole market on its own. As a rule, technologies requiring large investments of money and time (such as biotech medicines) will be licensed exclusively. Other technology (a research tool that requires little further development, for example) can be licensed widely and nonexclusively.
Research exemption
Sometimes an inventor becomes involved as a key scientific member of the team that further develops the ideas in the patent. For many inventors, however, particularly those in an academic setting, their involvement ends with filing the patent. Occasionally, an inventor will pursue development of their invention in a small start-up company that has licensed it from the academic institution. Alternatively, an inventor may simply continue to do follow-on research that is related to the original invention but not focused on its commercialization. In such cases, it is important for academic technology transfer professionals to consider how the researcher will be enabled to continue to conduct such research. For example, it is common for academic institutions in the United States to reserve the right for the continued use of a discovery for academic and nonprofit research purposes in any exclusive license to the patents pursued on that discovery.
In the European Union, the patent holder has the right to exclude others from using the patented invention. The owner of a product patent has the right to prohibit third parties from making, offering, putting on the market, using, possessing or importing the product. In the case of process patents, the patent protection extends not only to the application and offering of the protected process but also to such objects that are direct products of the protected process. The patent proprietor may transfer their proprietary rights in whole or in part to others by means of a license. An exclusive license means that no person or company other than the named licensee can exploit the relevant IP rights; the licensor is also excluded from exploiting the IP rights. If the licensor wishes to continue to conduct any activity covered by the IP (for example, a university licensor may wish to continue its research), or if the licensor has previously granted any rights in relation to the IP, the exclusive license will need to expressly state that it is exclusive subject to those carve-outs.
Under the European Patent Convention (EPC),
Under Swiss patent law, as another example, a similar exemption is granted. Pursuant to Article 9, paragraph 1, of the Swiss Federal Act on Patents for Inventions (Patent Act),
Reasons for patenting early research results
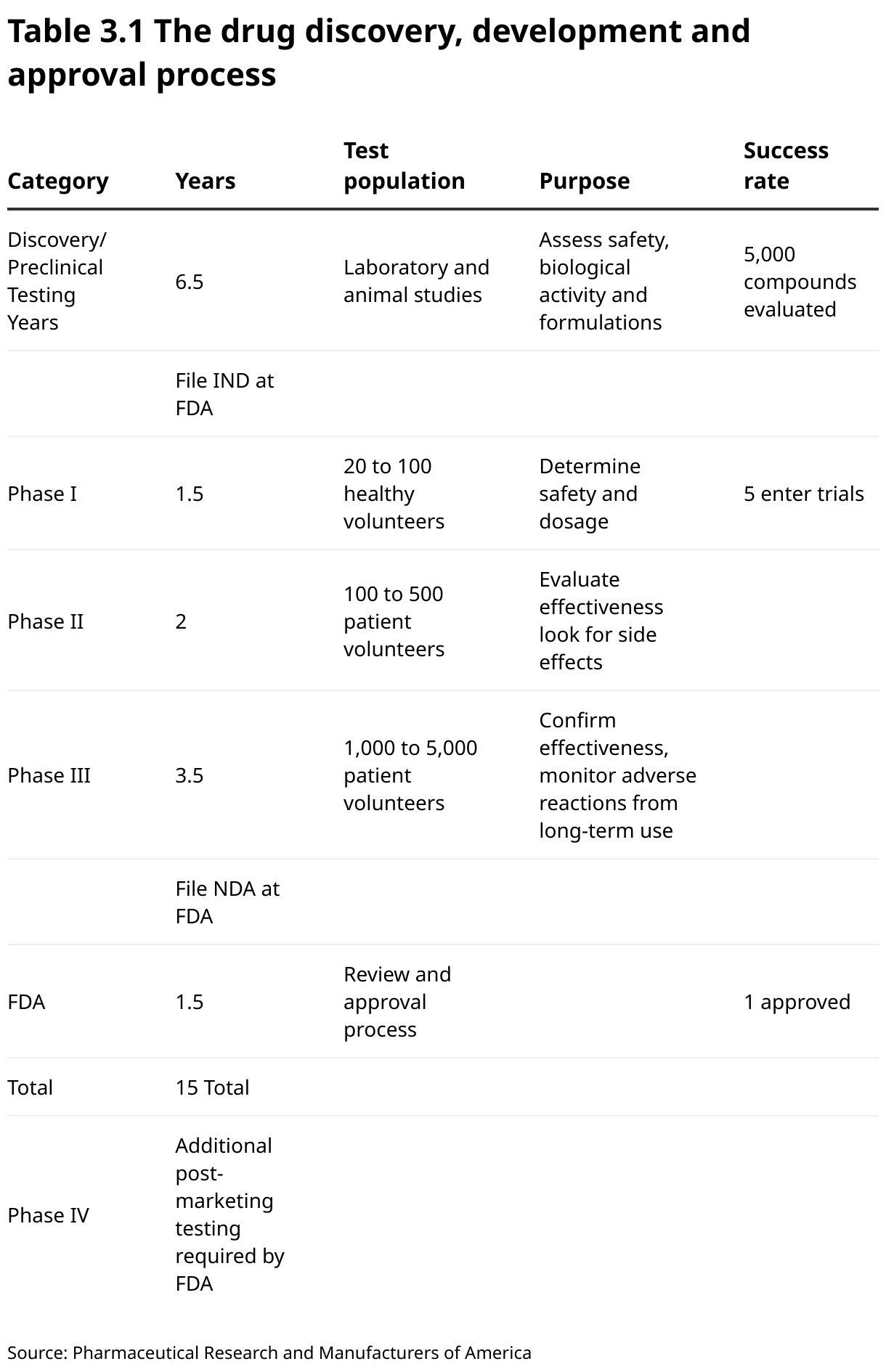
In private laboratories, research leading to a potentially commercially viable product is patented not only to protect the resulting product or technology, but also to attract investors and potential partners/collaborators. Whichever path is taken, this is the beginning of the journey for this unique discovery. Regardless of whether the invention is made in a public or private laboratory, because nearly all biotechnology products may affect either the health of humans or animals or the environment, commercialization is generally not possible without prior regulatory approval. Approval procedures can take many years. For example, it takes an average of 12–15 years to move a drug from the laboratory to the clinic or market. Only five in 5,000 compounds that enter pre-clinical testing make it to human testing. One of these five tested in people is approved. Table 3.1 outlines this process in its entirety from discovery and preclinical trials through the three phases of clinical trials for drug approval and on to further obligations, such as additional post-marketing testing required by the US Food and Drug Administration (USFDA) after the drug has been approved and is already on the market.
The processes shown in Table 3.1 and Figure 3.1 apply to the authorization of all medicinal products and not just to biotechnological drugs.
In the European Union, similar development and approval processes apply. These are shown in Figure 3.1.

Typically, once a patented invention has been licensed to a company (regardless of whether it is licensed from a public institution to a private company, or from a private laboratory to a private company), the company sets about refining the research and begins the development process. Through trial and error, the company gains the know-how necessary to produce a consistent product on a large scale. Each of these steps along the discovery spectrum requires funding, much of which is obtained through investment by the private sector.
Attracting financing
It is the early stages of development (proof of concept research) that are most vulnerable to perturbations in the capital markets. While it is relatively easy for entrepreneurs to obtain seed financing (generally from angel investors, governments and nonprofit foundations/organizations), it is the follow-on financing, such as the second and third rounds of venture investment required to fund companies beyond proof of concept and through clinical trials and approvals, that is often the most difficult to obtain. At this stage, for privately developed biotechnology products, what the capital markets require is the assurance that their investment in the discovery will bear a return on that investment. It is here that patents play a significant role (see “Academic institutions and the biotechnology industry” for detailed discussion of IP in financing). The patent portfolio associated with a particular product or technology can make or break investor confidence; therefore, throughout the development process, the company will seek additional IP to defend against competitors and to further buttress its portfolio. For example, a licensee will try to protect a key synthetic process; derivatives and variations of a chemical or biological structure; use of an important vaccine adjuvant; or a unique companion diagnostic.
Regulatory considerations for products of medical biotechnology
The United States of America
A crucial step in bringing a drug from bench to market is the regulatory review and approval process. Due to the significant US market, an application for drug approval is almost always filed with the USFDA.
The European Union
Regulatory review and approval processes in the European Union are generally within the competences of individual member states. According to Article 168 of the Treaty on the Functioning of the European Union there are a few exceptions. In these cases, the EMA, as an agency of the EU Commission, has the authority for regulatory review and market approvals. In the European Union there are several procedures available for the marketing authorization of medicinal products. The type of marketing authorization procedure depends on the medicine itself and whether the pharmaceutical company intends to market the medicinal product only in one EU Member State; in several EU member states; or in the European Economic Area (EEA). Medicinal products can be authorized by a national procedure, a mutual recognition procedure, a decentralized procedure or a centralized procedure. For the authorization of certain drugs, in particular medicines with new active substances for severe diseases, the centralized EU authorization procedure must be used. Innovative medicinal products and/or those where it is in the public health interest to do so, should be marketed throughout Europe.
Approval is granted by the EU Commission, which has devolved this task to the EMA, giving it responsibility for the scientific evaluation of applications for centralized marketing authorizations in the European Union. This authorization procedure allows pharmaceutical companies to submit a single marketing authorization application to the EMA and to market the medicine and make it available to patients and healthcare professionals throughout the EEA on the basis of that one marketing authorization.
Financing to complete regulatory review
One of the primary goals of fundraising in the biotechnology drug development process is to raise enough money to take the commercial invention through the regulatory review process. To obtain approval for biotech medicines, sponsors
For the purpose of illustration, this Primer will follow the path of drug discovery and approval through the USFDA. Figure 3.2 shows this pathway, which is discussed in more detail below.

Drug discovery/screening
R&D for new pharmaceutical products usually starts with a time-consuming and expensive screening process to pick out promising compounds or molecules from the vast number of natural and synthetic compounds available. Testing large numbers of compounds to see if they produce an appropriate biochemical or cellular effect is one of the first steps in the drug-discovery pathway for both chemical and biological drugs, including biotechnological pharmaceutical products.
Preclinical trials
Once it has been determined that a compound or molecule has therapeutic promise, the drug must be shown to have promise for use in humans. This phase, which bridges the gap between discovery and clinical trials, is generally considered the preclinical phase of drug development. It is at this point that the sponsor begins the process of gathering clinical data. In the United States, for example, sponsors are required to first file an investigational new drug (IND) application, which is a request from a clinical study sponsor for authorization from the USFDA to administer an investigational drug or biological product to humans.
The IND requires animal pharmacology and toxicology studies, manufacturing information and clinical protocols and researcher information
According to the Knowledge Network on Innovation and Access to Medicines (a project of the Global Health Centre at the Graduate Institute, Geneva, funded by a grant from the Open Society Foundations),
Clinical trials
In the United States, once the IND is approved by the USFDA, the sponsor sets out to gather data to file either a new drug application (NDA) under 505 (b)(1),
Clinical trials follow a rigorous protocol beginning with small-scale phase I studies, moving through slightly larger phase II trials to late-stage, large-scale phase III studies. Only if a treatment is successful in one phase, will it move on to the next. A successful clinical trial process continues until the sponsor files a marketing application (NDA or BLA) with the USFDA or a regulatory authority in another country for the medication to be approved for doctors to prescribe to patients.
The processes and the procedures of clinical trials in the European Union and EU member states are similar to those in the United States. All drug products, including biotechnological drugs, must go through preclinical and clinical trials before authorization. The phases and design of clinical trials may differ for certain diseases and specialized medicines, such as cancer drugs or gene therapies, but Box 3.2 provides a general overview of each phase of a clinical trial for most drugs.
Phase I to evaluate the safety of the drug candidate.
Between 20 and 80 healthy volunteers participate; the timeframe is variable but can take several months.
Phase II to assess effectiveness and further assess safety (e.g., side effects).
Administered to several hundred patients (depending on the drug) who have the associated disease or disorder; can take several months to years.
Phase III to test drug in large populations of patients.
Demonstrates whether a drug candidate offers a treatment benefit to a specific population and provides the basis for product labeling; this phase can last from one to four years.
Phase I
During phase I studies, researchers generally test a new drug candidate in between 20 and 80 healthy volunteers. The timeframe for this phase is variable but can take several months. The primary purpose of a phase I study is to evaluate the safety (for example, the side effects) of a new drug candidate before it proceeds to further clinical studies. In addition to safety, in a phase I trial researchers can answer questions related to how much of the drug is measured in the blood after administration, how the drug works in the body and the side effects associated with increased dosage.
Phase II
Around 70 percent of potential new drugs enter phase II. In phase II studies, researchers administer the drug to a larger group of patients (typically up to a few hundred) with the disease or condition for which it is being developed to initially assess its effectiveness and to further study its safety. A key focus of phase II studies is determining the optimal dose or doses of a drug candidate and how best to administer it to maximize possible benefits, while minimizing risks. The timeframe for this phase is also variable depending on the drug and can take several months to several years to complete.
Phase III
About 33 percent of drugs make it to phase III, which tests the potential treatment in the largest number of people. Phase III studies typically involve 300–3,000 participants from patient populations in which the medicine is eventually intended to be used. Phase III studies are often conducted as “double blind” studies: participants are assigned to receive the medication being evaluated or to a control group that receives either the current standard-of-care treatment or a placebo (a substance that has no therapeutic effect). Individual patients do not know to which group they have been allocated. In addition, the researchers do not know which patient group has received the drug. Phase III studies – among other things – demonstrate whether a drug candidate offers a treatment benefit to a specific population, provide more detailed safety data and serve as the basis for product labeling. Phase III trials last from one to four years.
When one or more phase III trials have been completed, the sponsor examines the results and decides whether the drug has demonstrated effectiveness and an acceptable safety profile in treating a disease. If so, the company can submit an NDA which contains all the data and information gathered at every stage of the process through to the results of the phase III clinical trial(s), as well as other information required by the applicable regulatory authority. The NDA is submitted to the USFDA (or analogous regulatory authorities outside the United States) for consideration for marketing approval.
According to the Knowledge Network on Innovation and Access to Health, this phase of the regulatory review process costs sponsors from hundreds of millions of dollars to more than USD 1 billion. A study published in the Journal of the American Medical Association
Post-approval and follow-up/pharmacovigilance
The sponsor remains involved after the product has been approved and the drug enters the post-approval phase of regulation, which includes post-market surveillance and the approval of any modifications that are made when the product is manufactured after approval. This phase can take several years and often decades. For example, in the case of cell and gene therapy products, which are required to sustain long-term effects on the body, post-approval follow-up can take up to 15 years.
Similar obligations also apply in the European Union and EU member states in the context of pharmacovigilance, which is defined by the EMA as the “science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other medicine-related problem”. In line with this general definition, the underlying objectives of pharmacovigilance in accordance with the applicable EU legislation are: (1) preventing harm from adverse reactions in humans arising from the use of authorized medicinal products within or outside the terms of their marketing authorization or from occupational exposure; and (2) promoting the safe and effective use of medicinal products, in particular through providing timely information about the safety of medicinal products to patients, healthcare professionals and the public. Pharmacovigilance is therefore an activity contributing to the protection of patients and public health.